14 案例系列3:自研平台
14.1 交大生信分析平台
14.1.1 学科、团队背景概况
生物信息学分析是交我算高性能计算平台的TOP需求(见图14.1、14.2)。自2022年以来,交我算已有324个医学账号,使用机时达6340万核时。在整个交我算资源使用中,医学院算力占比达14%,存储占比达40%,形成了交大版的”东数西算”————“医数交算”。
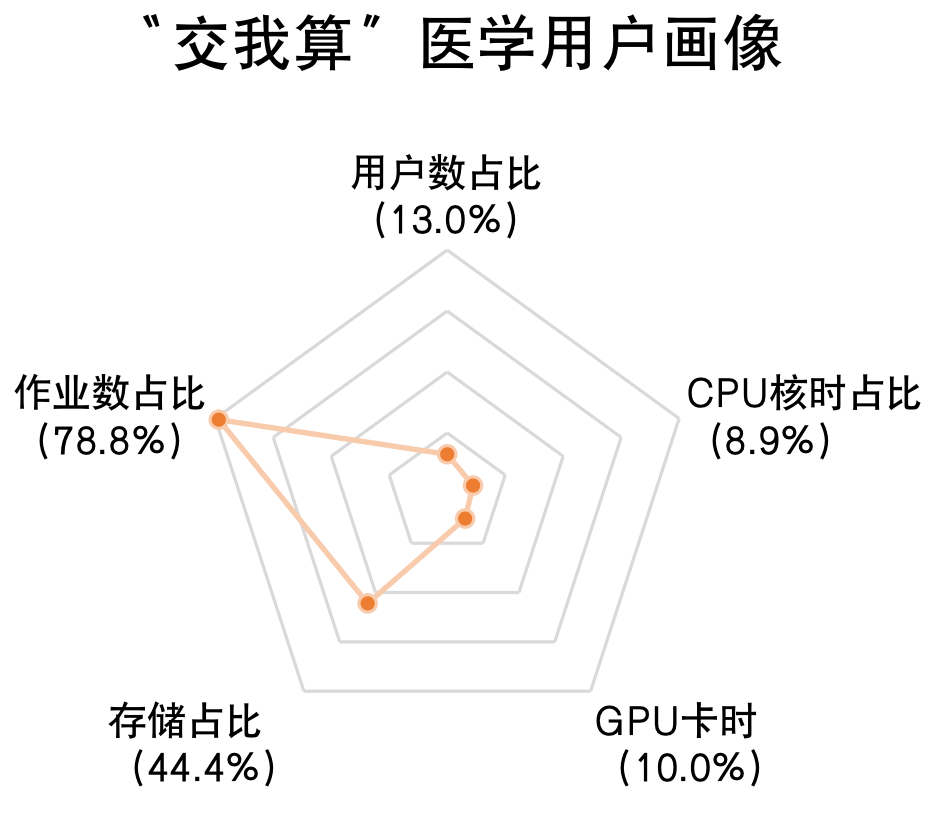
图14.1: “交我算”医学用户画像
图14.2: “交我算”学科核时分布
相比大规模并行计算应用的特点(例如,矩阵计算、格点计算、图计算等),即把一个大的计算问题通过数据划分,在数百或者数千个计算节点上并行计算。生物信息学中广泛使用的序列比对、序列组装、基因识别等组学分析计算,在计算模式上与大规模并行计算应用大相径庭,具有以下3个特征:
样本数据量大。以人类基因组测序为例,人类基因组有30亿个碱基对(约3G大小),而测序深度通常为30~50X,因此单个人类基因组测序样本的大小可达上百GB;人类的测序队列也在持续增长,从1990年的人类基因组计划[3],2008年的千人基因组计划[4],到2020年的中国代谢解析计划[5],类似的测序工作会产生海量的样本数据。
分析流程复杂。生信分析的计算过程往往是一个步骤间相互依赖的复杂流程,涉及到不同来源数据的相互整合或比较、不同软件的串联与组织等。
作业通量高。一次生信分析,通常是几十至上百样本同时运行,会有几百GB到几TB不等的中间数据产生、处理和存储。
生物信息学领域的计算应用,具备与传统并行计算应用完全不同的计算模式,如果沿用同一套计算框架,可能导致高性能计算平台的性能无法充分发挥,导致应用的计算性能显著下降。因此,为生物信息学领域,设计和构建一个面向高性能计算系统的专用领域应用架构,具有重要的意义。
14.1.2 平台的功能和创新特点
当前生物信息学分析面临着数据量大、分析复杂、分析通量高等诸多困难。为解决以上问题,近年生物信息学分析发展朝着分析标准化、可移植、可复现、并发高通量的方向发展,主要有3个方向:
采用多工具或多软件整合方式,如GATK4,其整合多个软件形成一个统一的多功能软件包,GATK最佳实践也是基因组分析的标准化流程。该方式在实际使用中存在软件安装困难、分析流程可移植性差、分析功能受限等问题。
采用通用工作流方式,其使用专门的工作流语言来构建分析流程,将分析过程对底层计算基础设施的要求与工作流描述分离开来。主流的工作流语言包括CWL、WDL、Nextflow、Snakemake等,表14.1对这几种主流的工作流语言进行了详细比较。该种方式使用门槛较高,需要分析人员具备流程开发、代码编写的能力。
采用组学相关多功能分析平台的方式,可提供一个个现成的分析工具或流程供用户使用,无需用户部署软件、编写流程。现有分析平台包括Galaxy、iMetaLab、nf-core、NASQAR等。
| 简称 | 发布时间 | 描述 | 优势 | 劣势 |
|---|---|---|---|---|
| CWL | 2014 | 一个用于描述和执行生物信息学工作流的开放标准 | 一个开放标准,具有广泛的社区支持和参与; 支持多种编程语言和工具,工作流描述和执行更灵活; 描述文件易阅读和理解,工作流的可维护性和可重复性高。 |
学习曲线较陡峭,描述文件复杂 |
| WDL | 2015 | 一种用于描述和执行生物信息学工作流的语言 | 一种简洁且易理解的语言,具有直观的语法和结构; 与其他生物信息学工具的集成性好,能方便地与现有工具交互; 可很好地支持并行和分布式计算,适用于大规模的数据分析。 |
社区规模较小,扩展性较差 |
| Nextflow | 2013 | 一个用于开发和执行可重复、可扩展的科学工作流的开源框架 | 支持多种编程语言和计算环境; 具有高度可扩展性和可重复性,能处理大规模和复杂的数据分析任务; 具有强大的错误处理和容错机制。 |
学习曲线较陡峭,描述文件复杂且冗长 |
| Snakemake | 2012 | 一个用于构建和执行可重复的数据分析工作流的Python库 | 具有直观的语法和易于理解的工作流描述文件; 支持并行和分布式计算,能处理大规模数据分析任务; 具有丰富的功能和生态系统,能与其他工具和平台进行集成。 |
扩展性较差,复杂流程的描述文件冗长且难以维护 |
Galaxy是由Blankenberg于2010年发布的最早的生物信息在线分析平台,其允许用户通过Web进行数据管理和基因组分析,经过不断的维护和更新,已成为全球使用最广泛的开源生信在线分析平台。iMetaLab是Liao等人于2018年开发的一个专用于宏蛋白质组学质谱数据处理和分析的Web平台。nf-core则是Ewels等人于2020年基于Nextflow工作流语言开发的新一代生信在线分析平台,其提供标准化的组学分析流程,同时支持Docker和Singularity等容器技术。NASQAR则是Yousif等人于2020年开发的使用R包为用户提供交互式数据分析和可视化的平台,可进行转录组数据预处理、RNA-Seq分析、宏基因组学分析和基因富集分析。
虽然上述研究提供了生物信息分析的在线使用平台,但仍存在许多问题,比如Galaxy存在流程组织复杂、定制困难;iMetaLab专注于蛋白组质谱单一领域;NASQAR聚焦于特定方向等问题。同时上述平台都未接入高性能计算系统,存储资源和计算能力存在较大的限制。
为应对生物信息学计算模式带来的挑战,并充分发挥高性能计算机的性能优势。交我算团队自主研发了一套面向高性能计算系统的生物信息学分析平台————交我算生信分析平台。平台具有用户登录、存储数据、筛选流程、运行作业等功能,兼具易用性和高效性,旨在帮助用户利用高性能计算集群开展生物信息学分析,高效实现高通量分析计算。
交我算生信分析平台(图14.3)是以页面可视化为核心的生物信息分析解决方案,帮助用户彻底摆脱纯代码界面操作的束缚,实现一键式自动化数据分析与报告交付。平台所有功能均可通过web界面可视化操作,用户无需配置Linux操作系统,安装复杂的分析软件,下载大量的数据库,只需在可视化界面中选择相应的分析流程,设置计算参数,即可在线进行生信分析,生成专业的分析报告。这大大的缩短了计算时间,能够做到快速、高效的交付实验结果。

图14.3: 交我算生信分析平台首页
平台主要有以下3个创新点:
整合了生物信息学领域的软件镜像库、分析流程引擎和参考数据集,提供可视化的一站式在线数据分析平台,提升了该领域计算的易用性;
开发了针对大量分析样本的批量提交功能,包含样本识别、目录识别、批量勾选,提升了高通量生物信息学分析的效率;
设计了一整套面向高性能计算系统的应用领域平台软件框架,为超算上领域应用平台的建设,提供了可借鉴的技术方案。
用户管理
为加强用户身份管理,平台要求用户登录jAccount完成账号注册和系统登录,系统默认用户名即其jAccount账号名。jAccount为上海交通大学统一身份认证系统,采用标准的基于OAuth2.0的OIDC身份认证协议。BIO-HPC通过授权码模式接入jAccount系统,获取用户必要信息,实现平台的用户管理,面向校内用户(包含校本部、医学院和附属院)提供计算服务。
数据管理
生物信息学分析有许多通用数据集,如各物种的参考基因组。为方便用户使用,免除重复下载操作,BIO-HPC下载了常用的生信分析参考数据集,存储至平台”公共数据”目录,供所有用户访问。BIO-HPC上已有的公共数据集如表14.2所示。
| 名称 | 描述 |
|---|---|
| AlphaFold2 | 预测蛋白质结构参考数据集 |
| dbsnp | 存储物种snp位点数据集 |
| Homo_sapiens.GRCh38 | 人类参考基因组GRCh38版本及注释 |
| Homo_sapiens.hg19 | 人类参考基因组hg19版本及注释 |
| mgy_clusters | MGnify蛋白质数据库相关数据集 |
| uniref90 | 全球蛋白质序列参考集 |
“个人数据”为用户自己上传或分析产生的私人数据,仅用户自己可见(图14.4)。页面允许用户进行目录/文件的重命名、移动、复制、下载和删除以及文件的上传和目录的新增。“识别样本”按钮用于识别出用户目录下的组学测序样本,“个人样本”可查看识别出的样本结果。
图14.4: 交我算生信分析平台个人数据页
流程管理
BIO-HPC最核心的功能,是提供了一系列标准的、易用的生物信息学分析流程。基于WDL通用流程语言,BIO-HPC封装好了常用的生信分析流程供用户使用,共65个,包括转录组分析流程、全外显子组分析流程等,其中使用率较高的如表14.3所示。
| 名称 | 描述 |
|---|---|
| basal_PE_RNA_seq | 双端测序数据RNA-seq基本分析 |
| MAGeCK_RRA_CRISPR | 基于模型的全基因组CRISPR-Cas9 敲除的分析计算流程 |
| CountSpacers_CRISPR | 对CRISPR结果中的spacer分布进行统计分析的流程 |
| ParallelFold | 基于 AlphaFold 进行高速蛋白质结构高精确度预测的分析流程 |
| AlphaFold_Multimer | 可并行处理多个fasta文件进行蛋白复合物结构模型预测的分析流程 |
用户在”公共流程”页(图14.5),可使用多种方式查询和筛选流程,可根据流程类别查询流程、可根据流程关键字模糊查询流程、可展示已收藏流程。对感兴趣流程,点击流程名可进入”流程详情”,查看流程的详细介绍,包括版本、使用次数、标签、描述、流程图、输入输出参数等;点击”启动”按钮即可新建作业;点击”星星”图标可收藏流程。
图14.5: 交我算生信分析平台公共流程页
作业运行
“新建作业”页面,用户可选择分析流程,设置个性化的流程参数,提交后运行作业。 用户可在”作业管理”页看到个人名下所有作业,包括”运行中”、“成功”、“失败”、“已取消”多种状态。运行中作业可终止;成功、失败或已取消作业可重复,即用相同参数快速提交一个新作业。作业名可点击跳转至作业详情页面,查看作业运行时间、状态、运行参数、作业结果等信息,“作业结果”可快速跳转至该作业结果目录。
14.1.3 平台的技术路线
交我算生信平台采用接入高性能计算系统,提供可视化分析界面,集成通用分析软件、流程和数据集的方式来应对生物信息分析存在的数据量大、分析复杂、分析通量高等问题。平台接入上海交通大学高性能计算系统,提供海量数据存储、批量数据分析的能力;平台采用WDL作为流程的组织方式,易学习、易迁移;平台使用Singularity作为流程软件的部署和调用方式,易复用、易迁移;平台做到数据存储、分析过程的可视化,上手简单、使用门槛低;平台集成通用的分析软件、流程和数据集,免除用户部署软件、编写流程、下载数据的繁琐工作,提升分析效率。
整体架构
交我算生信平台以上海交通大学校级高性能计算平台π2.0为基础平台(图14.6),采用客户端(浏览器)/服务端(高性能计算机)的模式构建。
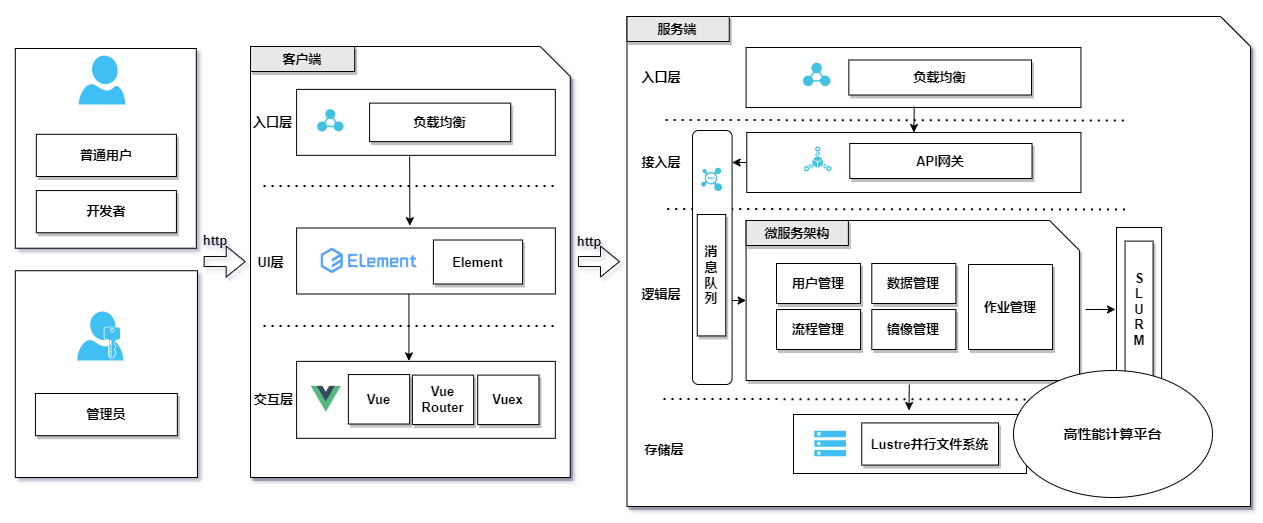
图14.6: 交我算生信分析平台架构设计图
服务端采用Python开发,使用微服务架构,提供平台的整体业务逻辑处理和数据存储能力。根据功能上的不同,拆分成用户管理、数据管理、流程管理、作业管理、镜像管理5个微服务模块。每个模块经统一API网关暴露出RESTful规范的API接口,供客户端调用。服务端入口配置有一个负载均衡,用于转发和代理客户端流量。
- 用户管理模块:提供对平台多角色、多租户并发访问的管理与控制,提供用户注册、审核、登录、密码重置等功能。平台将用户划分为3类角色:管理员、开发者、普通用户,分别赋予不同权限。管理员为具有最高权限的一个账户,系统交付时默认设置;开发者为流程开发人员,由管理员添加或赋权;普通用户为平台最终使用用户。模块涉及用户的登录和注册,对于用户登录,模块会通过JWT[17]进行身份校验,同时根据其角色进行接口限权;对于用户注册,会进行敏感信息加密和用户审核,保障安全。
- 数据管理模块:用于管理平台的公共数据和用户数据。公共数据提供常用的参考基因组、参考遗传变异数据库等,用户可共享使用,无需重复下载;用户数据会进行用户间数据隔离,保护隐私。模块通过计算文件md5、数据分片的方式,实现大文件断点续传和秒传的功能;基于正则匹配和计算汉明距离的方式,提供生物信息学分析常需要的”样本”功能,可自动识别同一样本的单双端测序文件并匹配相应样本名称。模块通过配置文件声明根目录的方式,接入高性能计算平台的并行文件系统提供数据存储能力。
- 流程管理模块:用于管理平台的分析流程,为公共流程和个人流程提供不同的处理逻辑。公共流程为管理员审核通过后、在平台上架的面向所有用户开放的流程;个人流程则是未上架的、由开发者用户在流程开发过程中临时保存的草稿。模块采用WDL通用流程语言作为流程组织方式,开发者用户可将生物信息分析过程通过WDL封装成具体的流程,在平台上架后供所有用户使用。
- 作业管理模块:用于处理用户作业的提交、运行、暂停和报告交付等逻辑。模块封装了Cromewell API[22]的SDK,可将作业提交至Cromwell,其负责对接高性能计算平台的作业调度系统Slurm,实现BIO-HPC调用高性能计算平台的计算资源。该模块还提供用户个性化参数设置、批量作业提交、并行化作业运行的能力。
- 镜像管理模块:用于上传与管理软件镜像,支持开发者上传Docker镜像和Singularity镜像,该模块根据文件名后缀判断镜像类型,并负责将Docker镜像转换成Singularity镜像,从而与WDL流程描述中声明的依赖软件进行绑定。平台已集成了许多生物信息学分析常用的软件镜像。
客户端为用户使用的界面,本平台为Web应用,因此是浏览器界面。客户端采用Vue3.2框架和ElementUI组件进行开发,集成TypeScript,支持i18n国际化、页面缓存、细粒度权限控制。客户端对平台所有功能进行了可视化包装,用户在浏览器上进行点击、输入等操作即可完成对生物信息学数据的存储和分析。对于不同的用户角色,客户端会展示不同的内容,因此客户端可分成3套界面:管理员界面、开发者界面、普通用户界面。
- 管理员界面:管理员可查看平台所有用户、公共数据、公共流程和所有镜像;可进行添加、激活或禁用用户,上传、删除或移动公共数据,审核、上架、下架公共流程,使用待审核流程运行作业等操作。
- 开发者界面:流程开发者可方便地在平台进行流程调试和发布,界面提供个人流程上传、描述、编辑、发布,使用个人流程运行作业等操作;同时支持开发者查询平台已有镜像或上传新镜像。
- 普通用户界面:提供了面向平台最终使用用户,即生物信息学数据分析人员的相关操作功能,包括上传、移动、删除、复制粘贴个人文件,搜索、运行公共流程,设置流程运行参数,取消运行作业,预览作业结果等功能。
所有界面都支持用户通过账号密码或上海交通大学统一身份认证系统jAccount登录平台,支持修改个人密码、个人信息等常见功能。
关键技术
考虑到协作开发的需求、当前互联网技术发展趋势、生信分析的大数据高通量特征、对接高性能计算系统的要求,BIO-HPC提出了以下几方面关键技术。
- 微服务开发
服务端采用了微服务开发的架构,该架构将项目拆分成多个单独的服务,做到高内聚、低耦合,非常适合协作开发的需求。BIO-HPC采用了Nameko作为Python微服务开发的底层框架,这是一款小巧、简洁、异步通信方式的微服务架构,其采用RabbitMQ消息队列作为消息中间件,使用生产者、消费者模式实现进程间通信,支持RPC、Restful协议接口。
BIO-HPC的服务端拆分成用户管理、数据管理、流程管理、作业管理、镜像管理5个微服务模块,每个微服务模块拥有其独立的进程、存储,单个微服务的故障对其它微服务的影响较小。微服务之上配备有API网关,其作为消费者与微服务进行通信。API网关是服务端API的提供者,采用Flask框架开发,使用Restful协议向客户端暴露接口;而API网关与微服务,微服务与微服务之间采用RPC协议进行通信。
- 日志追踪
享受了微服务架构带来的优势,也需要承担其带来的运维难度的提升。日志追踪是运维排障极其重要的一环,BIO-HPC存在复杂的进程间通信,客户端与API网关、API网关与微服务、微服务与微服务,复杂的进程通信和服务调用给日志追踪带来了比较大的难点。 基于此,BIO-HPC在客户端,给每个向服务端发送的HTTP请求,生成一个追踪id,即在HTTP协议头增加TraceId字段,该字段通过UUID方式生成。在服务端,进行以下改造:
① 设置一个当前线程的全局变量local_trace = threading.local(),该变量含有属性trace_id;
② API网关在接收到客户端的HTTP请求时,使用Flask的@app.before_request装饰器构造函数,将HTTP请求头中的TraceId值赋予local_trace,即local_trace.trace_id = request.headers[“TraceId”];
③ 改造Python的logging模块,嵌入全局local_trace.trace_id作为日志字段,从而可在logging的日志中打印trace_id字段;
④ 利用Monkey Patching方式动态修改Nameko的MethodProxy._call_函数,将local_trace.trace_id作为参数带入RPC通信的进程上下文worker_ctx;
⑤ 各微服务使用nameko.dependency_providers模块的DependencyProvider类构造一个子类LoggingDependency,在worker_setup函数中将进程上下文worker_ctx中携带的trace_id赋予相关日志句柄,其可在微服务中进行调用并打印日志。
基于以上操作,成功将客户端产生的TraceId传递到API网关和微服务,从而使客户端与API网关、API网关与微服务、微服务与微服务之间的调用透明化、可追踪化。
- 流程引擎
BIO-HPC以WDL为基础,设计了Web页面自动生成、参数自动解析的流程发布方式。开发者上传WDL文件后,平台首先进行文件格式的校验,然后解析出流程的输入参数及其格式,自动生成流程参数描述、流程参数编辑、作业参数输入等页面。开发者可在参数描述、参数编辑页面对参数做进一步修改,包括使用Markdown语言详细介绍该流程或某个参数,设置流程使用的参考基因组,添加参数的默认值、可选值等,还可设置某个参数是否需要展示给用户。基于这种方式,开发者可对一个流程设置多套参数模板,例如,若参考因组为人类基因组,则展示人源相关参数或设置相应默认值;如参考基因组为小鼠基因组,则展示小鼠相关参数或设置相应默认值。从而满足不同参考基因组、不同参数选择的多样性需求。
相应地,平台以Cromwell[22]工作流管理系统作为WDL的调度引擎,其可以将WDL描述的流程运行在CCI容器中。BIO-HPC通过作业管理模块与Cromwell进行交互,实现作业提交、运行、取消、查询等操作。Cromwell默认采用Docker容器作为软件的调用方式,同时作业运行在本地节点。因此,为了能接入高性能计算平台,BIO-HPC对Cromwell进行了一系列配置调整:采用Singularity作为软件的调用方式;接入Slurm作为后端的作业调度与运行系统;封装常用Slurm和Singularity运行参数作为WDL的runtime变量;通过ssh远程执行命令的形式,将作业经高性能计算平台的登录节点进行sbatch提交。经以上操作,作业管理模块成功将作业提交至高性能计算平台的计算节点运行。
- 批量提交
作业通量高是生信分析的典型特征之一,对应该需求,BIO-HPC提供了批量作业分析能力,为此平台实现了样本识别、目录识别、批量勾选3个功能。
样本识别:采用正则匹配方式,对用户目录下的文件名进行扫描,过滤出常用的组学测序文件,包括fastq、fq、fasta、fa、sam、bam、fastq.gz、fq.gz等格式文件。对过滤出的文件进行两两匹配,计算其汉明距离,若为1,则这两个文件属于一个样本的双端测序数据,即R1文件和R2文件;剩余未匹配到的文件则属于一个样本的单端测序数据。根据匹配结果,平台会提取出样本的样本名称。最终展示给用户的结果,包括样本名、R1文件、R2文件三个字段,结果经用户修改后提交,可存储至数据库。
目录识别:平台扩展了WDL功能,赋予了两个特权字段,分别为WDL输入参数中的String Path_$path和特定任务名的Task——task final_out。前者在平台上代表着输入参数为一个目录,用户在可视化输入参数时,可便捷地选择目录;后者声明了平台的最终输出文件或文件夹,包含两个固定输出参数final_out.out和final_out.out_dir。final_out.out声明需要输出的文件或文件夹,可为File|String,Array[File|String]格式;final_out.out_dir可选,用于声明输出文件的上级目录,可为String,Array[String]格式,其长度与final_out.out一致。 3)批量勾选:在客户端的作业提交界面,对于WDL中格式为Array[Array[File|String]]的输入参数,则判断其输入为多样本,用户可批量勾选识别出的样本;对于格式为Array[File|String]的输入参数,则判断其输入为多文件,用户可批量勾选个人目录下的文件;对于格式为String Path_$path的输入参数,则判断其输入为目录,用户可勾选单个目录。 结合WDL自身的scatter任务循环机制,平台可做到1次作业提交,批量作业并行分析的效果,包括:1)批量提交样本,对基因测序样本进行批量分析;2)批量提交文件,对文件进行批量分析;3)提交单个目录,对目录内的所有文件进行批量分析。以下为支持批量分析的一个WDL流程示例:
workflow test {
Array[Array[File]] readPairs
scatter (sample in readPairs) {
call echo { input: in1=sample }
call final_out {
input:
in1=sample[0], in2=echo.out
}
}
}
task echo {
\# in1为单个样本
\# 格式为:[sample_name, file1, file2]
Array[String] in1
command {
echo \${sep="," in1} \> sample.txt
}
output { File out = "sample.txt" }
}
task final_out {
String in1 \# smaple_name
File in2 \# echo.out
command { }
output {
String out_dir = in1,
File out = in2
}
}以上WDL代码构建了一个流程test,该流程输入参数为Array[Array[File]]格式,因此可输入批量样本;流程使用scatter机制对输入的样本循环运行任务task echo和task final_out。其中task echo将样本的3个字段:sample_name(样本名)、file1(R1文件)、file2(R2文件),拼接成一个逗号分割的字符串,输出到sample.txt文件;task final_out指示将task echo的输出文件sample.txt保存至目录sample_name下。
该流程执行后,在用户的作业结果目录下,会包含多个以样本名命名的文件夹,如sample_name1,sample_name2到sample_nameN,每个文件夹下都有一个sample.txt文件。
为提高批量作业的成功率,避免中间步骤出错导致的全部任务重新计算,BIO-HPC启用了Cromwell的缓存功能,即设置参数call-caching.enabled的值为true。该功能可对相同输入文件、相同参数的任务结果进行复用,只在失败任务处开始重新计算,从而极大地减少了失败作业的二次计算成本。
- 安全保障
为保障平台安全,BIO-HPC在用户系统和平台架构上建立了一系列安全保障机制。
用户密码:采用MD5和加盐的方式进行加密,保障密码安全。强制要求用户密码达到复杂性要求,用户修改密码时也会进行原密码校验,忘记密码时重置会进行邮箱、用户名和加密字段校验。
用户访问:采用JWT方式进行身份校验,JWT有效载荷只携带UID,不携带其它用户敏感字段。JWT签名通过加盐方式进行加密,在用户访问时进行校验。同时使用用户非敏感字段,采用交叉加密的方式形成一个SECRET字段,携带在JWT载荷中,用户访问时校验该字段。SECRET字段与JWT签名形成两层安全保障,攻击者破解JWT签名或SECRET字段两者中的任一者都无法成功伪造JWT。
服务端接口:每个接口都会进行参数校验,保证用户请求参数的合法性与安全性,对常见的SQL注入、越权请求都有进行判别。服务端每个接口,会进行详细的用户权限限制,无权限用户访问报错。
网络环境:平台运行在内网环境,通过Nginx的反向代理方式向外提供服务。Linux服务器防火墙只暴露必须的HTTP、SSH协议接口,不暴露其它接口;同时限制了服务器登录IP段,保证登录来源可信。平台在服务器上以普通身份运行,防止攻击者攻破平台后获取到Root权限。此外,平台设置了Nginx黑名单机制,执行脚本每小时定时分析Nginx日志,检测异常访问IP并禁用。
14.2 上海交通大学科学数据平台
14.2.1 科学数据平台简介
上海交通大学科学数据平台(scidata.sjtu.edu.cn,图14.7)秉持数据开放共享的初衷,致力打造校级科学数据全生命周期管理平台。
图14.7: 科学数据平台网站示意图
自国务院办公厅2018年发布《科学数据管理办法》(国办发〔2018〕17号)以来,科学数据的透明、开放、共享就一直是各省市科学数据中心的重要工作。《科学数据管理办法》强调加强和规范科学数据管理,保障科学数据安全,提高开放共享水平,对科学数据施行分级制度,积极开放共享非保密、不敏感的科学数据,推动国家科技创新、经济社会发展和国家安全。国际FORCE11组织也强调科学数据的FAIR原则,注重推动科学数据的可发现 (Findable)、可访问 (Accessible)、可互操作 (Interoperable)和可重用 (Reusable)。科学数据平台的初衷,即是加强校内科学数据开放共享,为全校科学研究提供助力。
科学数据从采集到发布,是一条完整的链路。“交我算”平台作为校级计算平台,解决了科学数据采集之后的存储、计算、分析过程。这些成果数据产生之后,科研团队面临的是数据整合、团队共享、成果发表等后续工作,以此让科学数据真正流通起来,发挥其应有的价值。科学数据平台作为数据管理平台,通过提供发布渠道、元数据记录、权限控制、数据链接等功能,意在解决科学数据的发布、共享、流通过程。两个平台功能互补,完善科学数据全生命周期的管理。
14.2.2 科学数据平台功能
数据发布
数据发布功能需要使用jAccount登录后使用,通过首页的“上传”功能按钮或个人账号旁的“加号”可以新建数据发布。平台支持多种类型的数据发布,包括但不限于数据集、成果图表、发表文章、专利、软件代码,用户可以根据自身需要选择相应的数据类型进行发布。每个数据条目关联的数据类型没有限制,可以是简单的文本文件,也可以是学科特异的数据类型,甚至是压缩包。
数据发布还提供丰富的元数据信息,通过选择数据类型,填写数据标题、描述、发布日期等可以编写数据条目的基本情况。创建者信息、贡献者信息、学科分类、基金支持、相关工作等补充信息,能够更加详细地记录数据条目的全方位信息,方便用户进行数据管理。
权限控制
平台提供数据条目和关联文件这2部分的权限控制,共3种模式:完全公开、元数据公开、完全私有,开放程度依次递减。(1)完全公开模式下,整个数据条目包含相关的文件都可以被其他用户或访客访问,适用于已经发表的科学研究成果,希望能开放共享给同行或感兴趣的科研人员使用,提升数据影响力及流通性;(2)元数据公开模式下,数据条目的相关文件无法被访问,其他信息不受限制,适用于成果发表初期,数据还无法完全共享的情况;(3)完全私有模式下,数据条目包含相关文件都只对发布数据的用户可见,其余用户无法访问,适用于研究进行阶段,需要对数据进行管理但还未达到数据发表要求。
团队空间
课题组负责人或管理人员可以建立专属的团队空间,上传团队展示图并简介团队信息。团队空间可以邀请已经在平台注册的人员加入,并根据情况对不同成员赋予3种不同身份:成员、编辑、管理员。在发布数据时选择指定团队空间,可以关联数据条目到此团队,此时,需要具有审核能力的编辑或管理员对数据进行审核,同意发布后才能正式发布。
所有属于此团队的用户,可以直接在团队空间首页查看并筛选所有只属于此团队的数据条目(图14.8),不论是公开数据或者是私有数据。团队空间可以方便地将还未正式发表的数据共享给团队中的所有成员,无需索要数据链接就能查看,数据上新时也可以及时同步。同时,管理员能够统一对数据进行管理,拥有审核、编辑的权限,方便筛查敏感数据或不宜发布的数据,及时制止。
图14.8: 科学数据平台数据集
投稿预审
针对已有成果产出但还未发表的数据,大多杂志在投稿时,要求将文章中涉及的数据内容放在公共数据库中,方便编辑审稿及之后的数据公开。科学数据平台提供数据查看链接,可以将私有的数据通过链接形式附在投稿文章中,提供给编辑查看。其他没有数据链接的人,则无法对数据进行查看。这种形式既能够满足投稿时的杂志要求,还能有效保护科研团队的权益,避免数据在发表前泄露。
数据动态
科学数据平台允许用户对数据的元数据、相关文件等进行更新,满足数据的动态变化需求。通过新版本生成,数据发布者可以对已经发布过的数据条目进行更新,修改相关的元数据信息,补充原先缺失的数据内容,或者增减关联的文件。这些信息变动,会自动记录在新的版本中,并且旧版本也不会丢失,所有历史版本都可以在数据详情页找到,方便数据发布者对数据变化进行追踪。
每个数据条目,还在详情页的右侧位置,展示了浏览量和下载量数据。通过这两项指标,可以清晰的观察到数据发布后,受到了多少的关注,以及数据的传播如何。数据发布者能够及时了解数据的统计信息,从而对数据质量、传播广度、影响力等做出综合判断。
14.2.3 科学数据平台建设方案
方案简介
科学数据平台建设,需要为科研团队提供用户友好的可视化界面,并且能与现有成熟平台的操作习惯一致,减轻用户的学习成本;同时,需要考虑全校科研团队的数据发布需求,部署能够承载未来3-5年数据量的存储空间。
因此,科学数据平台使用InvenioRDM开源技术方案,依托16.8PB的冷存储系统进行建设。InvenioRDM开源框架目前被国际数据发布平台ZENODO使用,由CERN团队开发维护,提供了数据存储、权限控制、前端页面等基础功能套件。使用InvenioRDM开源框架可以保持用户操作习惯,减少学习成本,同时利于后期个性化开发内容的增减。冷存储系统意在归档用户不常用或已经分析完成的数据,与需要发布的成果数据有重合,是科学数据平台存储对象的良好媒介。并且,冷存储系统的容量大,面对全校科研团队的数据发布需求也能够满足。
环境配置
服务需要运行在非ARM框架的Linux系统中,资源需求参考表14.4。
| CPU核数 | 4核 |
| 内存 | 8GB |
| 存储 | 服务存储 80GB,数据文件存储 30TB(根据需求量调整) |
部署科学数据平台服务需要预先在环境中安装依赖:
- 数据库:PostgreSQL 10+
- 搜索服务:OpenSearch (2.0+) 或 Elasticsearch (v7.0 - v7.10)
- 缓存及消息处理:Redis
- 主程序运行:Python 3.9,Docker 20.10.10+,Docker-Compose 1.17.0+
部署流程
- 安装InvenioRDM的管理包invenio-cli
在准备好的基础环境中,使用pip命令安装python包:pip install invenio-cli
可以使用如下命令确认安装完成及版本:invenio-cli –version
- 新建项目并填写相关信息
新建项目并初始化:invenio-cli init rdm -c v10.0
初始化过程中需要填写项目的相关信息,比如:项目名称、描述、项目管理人、联系方式、使用的数据库软件等。部分信息具有选项,会输出在命令行作为提示。
填写完相关信息后耐心等待初始化完成,会在当前路径下新生成一个以“项目名称”命名的文件夹,里面包含了所有项目运行时需要的文件。
- 以容器的形式运行服务
首先将当前版本项目运行需要的python依赖进行锁定:cd <project_name>; invenio-cli packages lock
接着,构建服务运行需要的镜像:invenio-cli containers build; invenio-cli containers setup
最后,将构建好的镜像运行成容器以启动服务:invenio-cli containers start
至此,服务会默认启动在https://127.0.0.1。如果需要更改服务运行的ip和端口,在项目文件夹中找到docker/nginx/conf.d/default.conf,修改服务的监听地址和端口即可。
14.2.4 科学数据平台建设现状
平台于2023年8月上线,对上海交通大学全校师生及附属机构提供服务。目前,平台已经协助上海交通大学医学院附属第九人民医院沈键锋教授发布了葡萄膜黑色素瘤中高频异常剪切异构体的蛋白结构数据(数据发表在scientific data杂志);将AlphaMissense的蛋白质预测数据同步到平台中(图14.9),提供给校内用户下载使用。
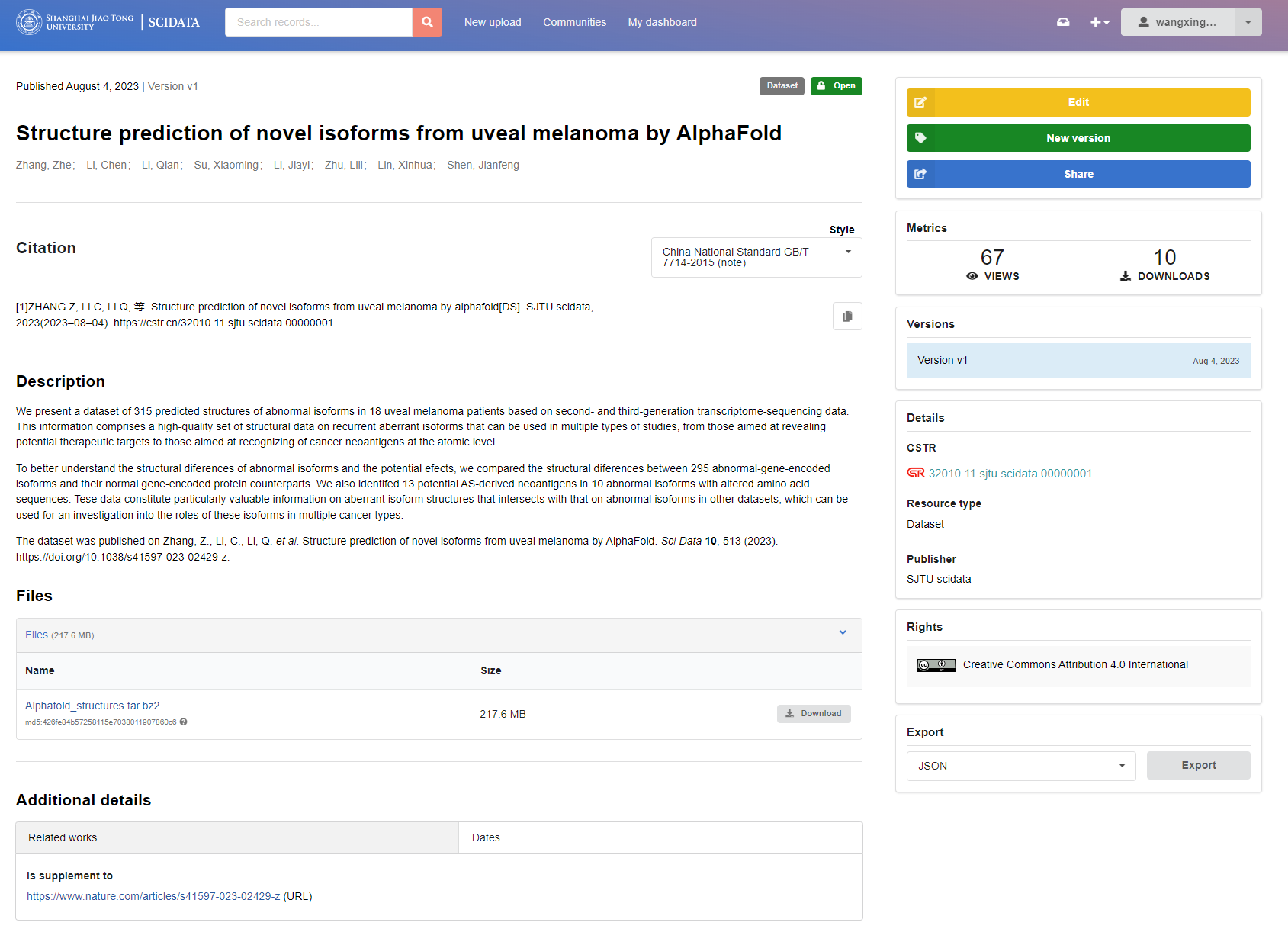
图14.9: 数据展示
公共数据资源部分,已经建立了4个公共数据集,共2.5T数据开放下载,将下载速度由外网下载的29小时,提升至25分钟,缩减了校内用户使用此类公共数据时的下载时间。平台用户注册面向全校开放,已收到2个校内课题组的申请,3个课题组正在洽谈中,未来希望能够为更多科研团队提供优质服务。
14.3 国产AI平台应用移植优化
14.3.1 国产昇腾平台背景
随着机器学习与生成式人工智能的飞速发展,从科研界到工业界对庞大算力的需求呈指数级增长。据统计,“交我算”的GPU资源占用率常位于95%以上,每个作业的平均排队时间为13小时,且50%左右的作业与人工智能相关。因此,“交我算”作为校级计算平台,需要建设更高性能的大型算力基础设施,以满足用户日益增长的智能计算需求。
多年以来,英伟达的GPU芯片以其Tensor Core技术、高带宽内存以及强大的多GPU互联能力,在矩阵计算和并行计算上具有突出的性能,被广泛认为是人工智能领域领域的首选计算平台,PyTorch、Tensorflow等主流的深度学习框架也主要基于英伟达GPU开发,具有垄断级别的地位。而随着国际形势变得错综复杂,中美贸易冲突不断加剧,美国不断升级停止向中国出口高端GPU芯片的禁令。根据最新规定,从2023年11月16日开始,英伟达将无法向中国出口适用于AI和HPC计算的A100、A800、H100、H800、L40、L40S以及RTX4090显卡。
为应对技术封锁的“卡脖子”手段,华为、海光、寒武纪等国内科技公司也在GPU、DSA(Domain Specific Architecture)芯片的设计研发与生产上投入多年,并取得了显著进展,不仅在性能上与国际主流产品逐渐缩小差距,而且在能效比、算力密度等方面展现出独特的优势。这不仅代表了我国在半导体技术领域的一个重要突破,也为国内外的科研和工业应用提供了新的动力和可能。
华为推出了其自主研发的专用于AI领域深度神经网络训练任务的昇腾910系列处理器,以其独特设计的达芬奇架构,在FP16和INT8算力上具有明显优势。2023年,华为发布了新一代昇腾处理器,智能算力大幅提升。以面向训练场景的昇腾910B加速卡为例,能够提供峰值达到313TFLOPS的半精度浮点算力,达到英伟达A100的水平。与GPU相比,昇腾处理器具备以下特点:
- 强大的较低精度算力,为深度神经网络设计多个专用计算单元,对神经网络计算进行了硬件级优化。
- 独特的深度学习软件栈,建立基于自研硬件的原生软件生态。
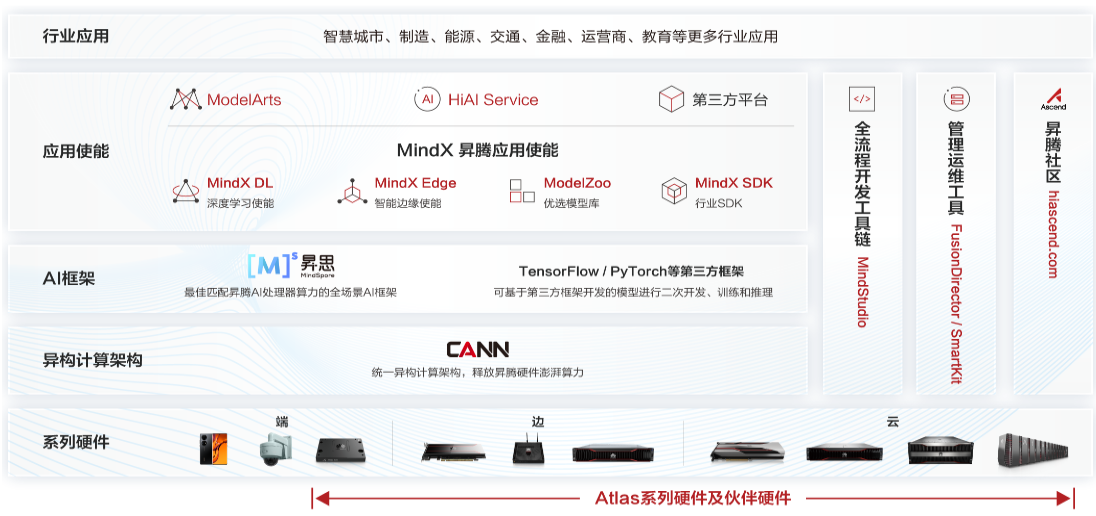
图14.10: 昇腾AI软硬件平台
“交我算”与华为昇腾合作,在校内搭建国产AI测试平台(图14.10,表14.5),邀请校内用户上线试用,并筛选不同方向的重点用户AI应用移植到昇腾平台,对比训练性能,探索建设高性能数据中心国产解决方案的可行性。
| 1 × Atlas 800 9000 训练服务器 | |
|---|---|
| CPU | 4 × HUAWEI Kunpeng 920 5250 (2.6GHz, 48 cores) |
| NPU | 8 × Ascend 910A 32GB |
| 架构 | Arm |
| 2 × Atlas 800T A2 训练服务器 | |
|---|---|
| CPU | 4 × HUAWEI Kunpeng 920 5250 (2.6GHz, 48 cores) |
| NPU | 8 × Ascend 910B3 64GB |
| 架构 | Arm |
14.3.2 移植方法
在昇腾上,深度学习的计算过程主要由CANN (Compute Architecture for Neural Networks)软件栈控制,类似GPU上的CUDA软件,允许开发者使用Ascend C语言从硬件底层进行软件开发。对于深度学习框架,CANN对PyTorch和TensorFlow进行了深度适配,提供了比较方便的自动迁移工具,能够使用户在不改变原来编程习惯的前提下,移植到昇腾上训练神经网络模型。另外,华为自研MindSpore框架也同时支持昇腾NPU和英伟达GPU,用户能够自行从原来的框架迁移到MindSpore上开发,以获取更好的性能。
华为还提供了开源模型库ModelZoo,由官方和合作者共同维护,其中包含大部分已经完成移植优化的计算机视觉、自然语言处理等领域的经典模型,如ResNet50、Unet、BERT等。
将人工智能应用移植到昇腾上运行和后续开发具备一定挑战,主要由以下几点: 1. 使用的软件栈与GPU不同,具备一定的学习成本。 2. 直接使用自动迁移方法会造成性能损失,需要进一步的性能精度调优,或直接采用ModelZoo中的相同模型代码实现。 4. 平台生态尚不完善,可能存在少量PyTorch算子未适配,需要在昇腾开源社区提交issue求助或自行实现。
模型迁移流程如图??中所示,以下将以PyTorch网络模型迁移为例,具体介绍将GPU上的AI应用移植到昇腾上的方法和技巧。
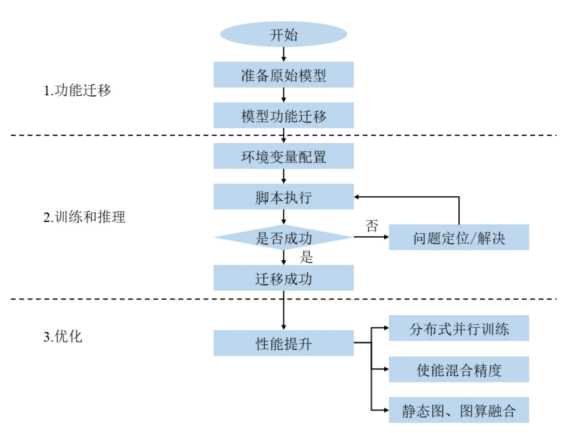
图14.11: 模型迁移流程
环境准备
- 在昇腾集群上部署CANN异构计算架构软件包,用于支持代码开发和编译,以及应用程序运行或进行训练脚本的迁移、开发和调试。
- 安装对应版本的kernel二进制算子包。
- 安装PyTorch框架,包括官方PyTorch软件与对应昇腾PyTorch Aadapter插件的安装。目前CANN 7版本支持PyTorch 1.11、PyTorch 2.0.1和PyTorch 2.1版本,能覆盖大多数PyTorch项目。
模型迁移
模型能否迁移主要取决于模型算子是否支持昇腾处理器,因此需要首先准备GPU版本的训练脚本,运行昇腾提供的PyTorch Analyse迁移分析工具,该工具会扫描脚本中的API,统计算子支持情况并输出为csv文件,如果不支持算子列表为空,即可开始模型移植。
./pytorch_analyse.sh \
-i ~/path/to/model/train.py \ #待分析脚本路径
-o ~/path/to/result/ #分析结果输出路径采用自动迁移方法,首先配置环境变量
export PYTHONPATH=/usr/local/Ascend/ascend-toolkit/latest/tools/ms_fmk_transplt/torch_npu_bridge:$PYTHONPATH然后在训练脚本中导入昇腾PyTorch库代码
import torch
import torch_npu
import transfer_to_npu运行训练脚本,其中的算子会通过接口自动转换为昇腾适配的NPU算子,观察输出文件和loss曲线等,检查模型迁移训练结果。
对于多卡任务,由于昇腾不支持DataParallel场景的并行,需要基于DistributedDataParallel场景修改单卡脚本为多卡脚本,并替换原生nccl为hccl (华为集合通信库),才能在昇腾上进行训练。
性能调优
通常情况下,实现自动迁移,模型训练性能仅能达到A100的20%~50%,如果要进一步提升昇腾上的训练性能,需要采取一些调优手段,针对NPU特性进行部分优化。以下是一些常用的调优方法:
- 替换ModelZoo模型
ModelZoo中提供了许多已经在昇腾上优化过的典型神经网络的完整代码,可以从中选取对应网络,替换数据集和数据预处理以适配待移植项目,能够得到较好的性能。 ModelZoo链接:https://gitee.com/ascend/modelzoo
- 自动混合精度 (AMP)
混合精度训练是在训练过程中使用单精度 (Float32) 与半精度 (Float 16) 数据类型,两者结合并使用相同的超参数,实现近似单精度的水平。由于昇腾处理器的架构特性,自动混合精度能够显著提高大部分模型的训练性能,但是也有可能引入误差和溢出错误,需要结合动态损失缩放实现。
- 算子二进制调优
PyTorch框架提供与算子编译相关的二进制配置参数,可以配置模型编译时优先查找当前编译好的算子二进制文件,若不存在再进行在线编译,能够减少编译时间,提高模型训练性能。参数配置代码如下:
- NPU亲和优化器替换
使用昇腾AI处理器亲和的融合优化器替换原生优化器,能够将连续多个小算子融合后在CPU上下发,提高模型训练的性能。目前常用的SGD、Adam、AdamW等优化器均提供了对应的NPU亲和版本供替换。
- 分析性能瓶颈
Ascend PyTorch Profiler是针对PyTorch框架开发的性能数据采集和解析工具,通过在PyTorch训练脚本中插入Ascend PyTorch Profiler接口,执行训练的同时采集性能数据,完成训练后直接输出可视化的性能数据文件,提升了性能分析效率。Ascend PyTorch Profiler接口可全面采集PyTorch训练场景下的性能数据,主要包括PyTorch层算子信息、CANN层算子信息、底层NPU算子信息、以及算子内存占用信息等,可以全方位分析PyTorch训练时的性能状态。针对某个算子出现的性能瓶颈,可以提交算子优化需求到昇腾社区。
- AOE调优
利用AOE (Ascend Optimization Engine) 自动调优工具,充分释放有限的硬件资源性能,以满足算子和整网的性能需求。根据dump算子图文件,通过生成调优策略、编译和在运行环境上验证的闭环反馈机制,不断迭代出更好的调优策略、最终达到最佳的训练性能。
14.3.3 移植案例 - 基于Unet的癌症组织图像分割模型训练
癌症的病理诊断是肿瘤治疗中至关重要的环节,它关乎治疗方案的制定、预后的评估以及临床决策的制定。在这一过程中,基于组织学的肿瘤特征评估被视为金标准。肿瘤组织的·苏木素-伊红(hematoxylin-eosin staining, H&E)染色图像能够揭示细胞核和细胞质的形态特征,为医生提供足够的信息来判断肿瘤细胞的良性或恶性。阅读这样的医学图像需要临床医师经过多年的培训与实践,利用其专业知识与丰富经验。但由于病变细胞在形态上的多样性和微观图像的复杂性,以及患者个体的差异性,完全依赖医师主观经验经验的阅片可能导致诊断结果的不确定性。
本案例基于PyTorch框架实现,设计了一个基于UNet的神经网络结构用于训练。UNet网络因其在医学图像分割中的出色表现而被广泛采用。它的对称结构包括一个收缩路径,用于捕获图像内容,和一个对称的扩张路径,用于精确定位感兴趣的区域。这种设计使UNet能够在不同的分辨率上捕捉图像特征,并通过跳跃连接将这些特征传递到网络的更深层,从而在上采样过程中恢复详细的空间信息。
交我算团队实现了将本项目从GPU移植到昇腾上完成单卡训练和多卡训练,采用了自动混合精度、算子二进制调优、NPU亲和优化器替换等调优策略提升训练性能。测试结果如图??所示,调优后910B上的单卡训练性能能够达到英伟达A100的62.4%,多卡训练性能达到A100的66.6%。
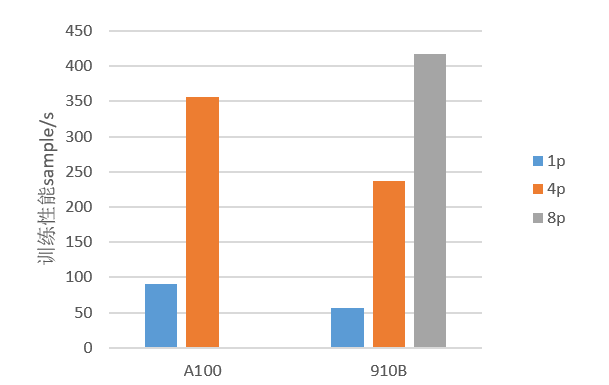
图14.12: 昇腾性能对比